Installation¶
Prerequisites¶
Python 3.11+
A UNIX-like environment (e.g. MacOS, WSL, Ubuntu)
A recent version of PostgreSQL (ideally at least 11+)
A modern Java runtime (if using DynamoDB for the Gene Normalizer database)
Library installation¶
Install FUSOR from PyPI:
pip install fusor
Data setup¶
Universal Transcript Archive (UTA)¶
The UTA is a dataset of genome-transcript aligned data supplied as a PostgreSQL database. Access in FUSOR is supplied by way of Cool-Seq-Tool; see the Cool-Seq-Tool UTA docs for some opinionated setup instructions, or you can use Docker to setup (see Docker for more information).
At runtime, UTA connection information can be relayed to FUSOR (by way of Cool-Seq-Tool) either as an initialization argument or via the environment variable UTA_DB_URL. By default, it is set to postgresql://uta_admin:uta@localhost:5432/uta/uta_20241220 (postgresql://<user>:<password>@<host>:<port>/<database>/<schema>.
SeqRepo¶
FUSOR relies on Seqrepo, which you must download yourself.
FUSOR uses Seqrepo to retrieve sequences at given positions on a transcript.
From the root directory:
pip install seqrepo
sudo mkdir /usr/local/share/seqrepo
sudo chown $USER /usr/local/share/seqrepo
seqrepo pull -i 2024-12-20/ # Replace with latest version using `seqrepo list-remote-instances` if outdated
If you get an error similar to the one below:
PermissionError: [Error 13] Permission denied: '/usr/local/share/seqrepo/2024-12-20/._fkuefgd' -> '/usr/local/share/seqrepo/2024-12-20/'
You will want to do the following: (_Might not be ._fkuefgd, so replace with your error message path_)
sudo mv /usr/local/share/seqrepo/2024-12-20._fkuefgd /usr/local/share/seqrepo/2024-12-20
exit
Use the SEQREPO_ROOT_DIR environment variable to set the path of an already existing SeqRepo directory. The default is /usr/local/share/seqrepo/latest.
Gene Normalizer¶
Finally, FUSOR uses the Gene Normalizer to ground gene terms. See the Gene Normalizer documentation for setup instructions.
Connection information for the normalizer database can be set using the environment variable GENE_NORM_DB_URL. See the Gene Normalizer docs for more information on connection configuration.
As a default, this connects to port 8000: http://localhost:8000.
Docker¶
FUSOR’s dependencies can be installed using a Docker container.
Important
This section assumes you have a local
SeqRepo
installed at /usr/local/share/seqrepo/2024-12-20.
If you have it installed elsewhere, please add a
SEQREPO_ROOT_DIR environment variable in .env.shared.
You must download uta_20241220.pgd.gz from <https://dl.biocommons.org/uta/> using a web browser and move it to the root of the repository.
If you’re using Docker Desktop, you must go to
Settings → Resources → File sharing and add
/usr/local/share/seqrepo under the Virtual file shares
section. Otherwise, you will get the following error:
OSError: Unable to open SeqRepo directory /usr/local/share/seqrepo/2024-12-20
To build, (re)create, and start containers:
docker volume create uta_vol
docker compose up
Tip
If you want a clean slate, run docker compose down -v to remove
containers and volumes, then run
docker compose up --build to rebuild and start fresh containers.
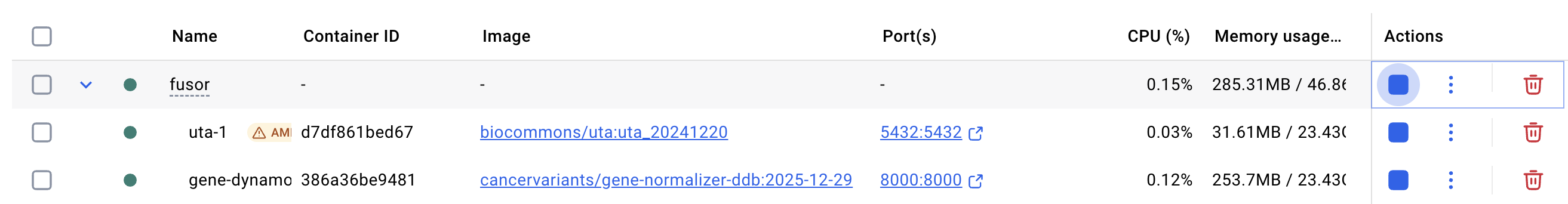
In Docker Desktop, you should see the following for a successful setup:
Note
python-dotenv can be used
to load environment variables needed for analysis notebooks in the
notebooks directory. Environment variables can be found in
.env.shared.
Check data availability¶
Use the fusor.tools.check_data_resources() method to verify that all data dependencies are available:
>>> from fusor.tools import check_data_resources
>>> status = await check_data_resources()
>>> assert all(status) # passes if all resources can be acquired successfully